Esbozada una imagen de la materia, desde los cuerpos macroscópicos hasta las partículas elementales, vamos a aplicar nuestras ideas para entender a los sólidos y en particular a los sólidos cristalinos, y poder contestar así las preguntas que hicimos al principio. Para ello formularemos la teoría atómica del sólido, con la que se trata de entender las propiedades de los cristales en base a las de los átomos que lo forman y a la manera en que ellos interactúan, aplicando siempre la mecánica cuántica. Otra vez tenemos todos los elementos de la teoría de un sistema físico: la descripción de los subsistemas, la interacción entre éstos y las leyes de movimiento. De ahí extraeremos conclusiones que luego habrán de verificarse experimentalmente, con la consecuente retroalimentación a la teoría. Esta es la espiral sin fin de la física. Un sólido cristalino está formado por un arreglo ordenado de átomos. Este arreglo o red cristalina puede generarse repitiendo indefinidamente un arreglo elemental, llamado celda unidad. De esta forma se obtiene una estructura periódica que permanece invariante frente a unas ciertas translaciones, como las grecas de Mitla.
Podemos imaginarnos una infinidad de celdas unidad y, por tanto, de grecas o redes cristalinas resultantes. Sin embargo, las matemáticas han puesto cortapisas a nuestra imaginación pues el requisito de invariancia translacional es fuerte. Por ejemplo, las celdas unidad no pueden ser de forma pentagonal, ya que con pentágonos no es posible cubrir un plano totalmente sin dejar resquicios. Los geómetras han encontrado que sólo unos cuantos cientos de estructuras cristalinas diferentes son posibles y los han clasificado minuciosamente. Aunque ya los griegos habían hecho observaciones sobre los cristales, su estudio se inicia en el siglo XVII, por Stensen y Guglielmini, llegándose a encontrar en 1772 una ley enunciada por Romé de l'Isle: en todos los cristales de la misma substancia, los ángulos entre caras correspondientes tienen el mismo valor (cuando se les mide a igual temperatura). De l'Isle no logró proponer un modelo que diera cuenta de su observación y fue René JustHauy, al dejar caer un cristal de calcita y observar las caras idénticas de los fragmentos, quien sugirió la existencia de la celda unidad a la que llamó molécula integrante. Él empleaba sólo tres tipos distintos de moléculas integrantes pero, como ya lo mencionamos, se tienen muchos más como demostró Bravais el siglo pasado.
TEORÍA ATÓMICA DE LOS CRISTALES
Desde el punto de vista físico, cuál de esas estructuras cristalinas ha de tomar el sólido depende de varios factores, siendo los principales el tamaño de los átomos que lo forman y el tipo de fuerzas que actúa entre ellos. En función de estas últimas, y en completa analogía con el caso molecular, se clasifican los cristales en iónicos, covalentes o metálicos. Veamos algunas de sus características. Los cristales iónicos se enlazan con fuerzas interatómicas del tipo de Coulomb. Cuando un átomo con un electrón fuera de capa cerrada se encuentra con otro que requiere un electrón para completar la capa, el primero cede al electrón tan gustosamente como el otro lo atrapa. Pero entonces se forman dos iones, uno positivo porque cedió una carga negativa y el otro negativo porque adquirió un electrón adicional. Por lo tanto, se atraen eléctricamente con una fuerza omnidireccional, para la cual no existe dirección privilegiada. La descripción que hemos hecho se aplica a los átomos alcalinos frente a los halógenos. De hecho, los cristales iónicos por excelencia son los halogenuros alcalinos, siendo el más conocido el cloruro de sodio, la sal común.
A diferencia de los cristales iónicos, en que los electrones se reparten en los iones, en los cristales covalentes los electrones se comparten. La nube electrónica se dispone de manera tal que los iones positivos son atraídos hacia los sitios en que aquélla se localiza principalmente. Estos sitios preferenciales no son cualesquiera, por lo que el enlace covalente se da sólo en ciertas direcciones. Estos cristales covalentes surgen de elementos como el germanio, el silicio y el carbono. Por ejemplo, el carbono tiene cuatro electrones en su capa más externa, que se llena con ocho. Por lo tanto, al átomo de este elemento le da igual, por decirlo así, ceder sus electrones o aceptar otro número igual para llenar su capa y llegar a un estado de menor energía. Entonces, decide compartir sus cuatro electrones dando origen con un enlace covalente a un cristal tetraédrico.
Los cristales metálicos, finalmente, se dan cuando los átomos que los forman tienen electrones muy poco amarrados que con poca energía se liberan de su ión y prefieren deambular por todo el cristal. El origen de esta energía y en consecuencia la existencia del enlace metálico, se puede explicar con base en la mecánica cuántica y su principio de incertidumbre. Recordemos que este principio nos dice que no es posible definir al mismo tiempo la posición y la velocidad de una partícula. Entonces, mientras mayores sean las limitaciones espaciales a las cuales se restringe una partícula, también será más grande la incertidumbre en la velocidad, y por tanto la velocidad misma de la partícula. En otros términos, mientras menos intentemos localizar a un electrón, menor podrá ser su velocidad y, por lo tanto, su energía cinética. Esta disminución en energía que obtiene el electrón al moverse en todo el cristal y no dentro de un átomo, puede ser suficiente para compensar la energía de amarre al átomo. Esto sucede con los metales, en los cuales existe un gas de electrones, e inmersos en él los iones positivos. Estos últimos se atraen unos a los otros por medio del gas de electrones.
ALGUNAS PROPIEDADES DE LOS CRISTALES
Con la imagen que hemos construido de los tres tipos de cristales, ¿qué propiedades podemos esperar de cada uno? En el metal, pletórico de electrones itinerantes, casi libres, esperaríamos que un campo eléctrico pueda mover fácilmente a esas partículas de valencia no amarradas a ningún átomo en particular. Pero esto es lo que caracteriza a un buen conductor eléctrico. Por otro lado, cuando incide un rayo de luz sobre el cristal metálico, los campos electromagnéticos que forman la onda luminosa hacen oscilar a los electrones libres, que generan al acelerarse una radiación reflejada. Por lo tanto, conducen mal la luz al ser buenos reflectores: en otros términos, son opacos. En cuanto a sus propiedades térmicas, podemos esperar que los metales sean buenos conductores del calor, siendo la explicación microscópica semejante: si se aumenta la temperatura en un cierto punto del metal, esto implica aumentar la energía cinética de los electrones menos amarrados, que luego transportan esa energía a todos los confines del cristal. Por otro lado, el amarre metálico proveniente del gas de electrones es poco direccional. Los átomos de la red pueden desplazarse de su posición de mínima energía sin que ésta se altere mucho, ya que el gas de electrones, por decirlo así, se adapta a las nuevas posiciones de los átomos. Esto implica, claro, que los metales se pueden deformar sin llegar a la fractura. Finalmente, y otra vez debido a que el amarre entre iones procede del muy dúctil y poco exigente gas de electrones, se pueden dar muy diversas mezclas de dos metales, sin que dependan fuertemente de las proporciones relativas de cada elemento. De ahí que exista una gran variedad de aleaciones metálicas.
Pensemos ahora en la imagen nuestra de un cristal covalente, en que los electrones de valencia están ocupados en ligar al cristal. No hay en este caso quien transporte la carga o la energía térmica. Si no hay agentes portadores, la información no llega nunca, o al menos tarda mucho en llegar. Resultado neto, los cristales covalentes no son buenos conductores ni de la electricidad ni térmicos: se pueden usar como aislantes. Como dijimos antes, el enlace covalente es muy direccional y por tanto la posición de equilibrio en la red puede ser delicada. Estos sólidos, en consecuencia, no serán dúctiles sino quebradizos. Finalmente, no existe el mecanismo por el cual se reflejaba la luz al incidir en un metal, y podemos esperar que los cristales covalentes sean buenos conductores de la radiación electromagnética, o sea transparente.
Propiedades muy semejantes podemos esperar de los metales iónicos, si acaso alguna diferencia en sus propiedades ópticas y mecánicas. Por ejemplo, cuando incide sobre un cristal iónico una onda electromagnética cuya longitud es mucho mayor que la distancia típica que separa a los iones, tanto el ion positivo como el negativo sienten un campo eléctrico del mismo signo. Pero responden a él de manera contraria y se puede excitar una vibración en la malla cristalina si la frecuencia es la apropiada. A esta frecuencia, el cristal iónico absorbe la luz y ya no es transparente. ¿Quién no ha sentido frío en la mano al tocar una perilla metálica? ¿Quién no ha visto los destellos primorosos de los diamantes? ¿No sabemos que los mejores espejos están metalizados? ¿O que las extensiones eléctricas son en general de cobre, un metal? Todo ello se explica con los modelos para los sólidos que hemos desarrollado.
EL MODELO DE PARTÍCULAS INDEPENDIENTES
Así como en el átomo generamos un modelo de capas, en que sólo se toma en cuenta la atracción que ejerce el núcleo sobre cada electrón despreciando la interacción entre éstos; en la misma forma que para el núcleo inventamos un modelo de capas en que la parte principal de la interacción entre los nucleones se agota en producir una fuerza promedio; así también para los sólidos se ha desarrollado un modelo de partículas independientes que en este caso se llama la teoría de bandas. E igual que en la física atómica y en la teoría nuclear, este modelo resulta esencial para aplicar las ideas cuánticas al estudio de los cristales. Para entender la teoría de bandas, es conveniente imaginarse que formamos el cristal juntando un átomo tras otro. Por simplicidad supongamos que todos los átomos son iguales. Cuando los átomos están muy alejados unos de los otros, las energías de los electrones son idénticas a las del átomo aislado. Cuando los acercamos, la presencia de un átomo perturba un poco los niveles de energía del otro. En consecuencia, de cada nivel atómico se genera una banda de niveles, tan próximos entre sí que forman un continuo. En alguno de estos niveles de energía estarán los electrones del sólido.
Dependiendo de los átomos de que se trate, las bandas provenientes de niveles atómicos contiguos pueden superponerse o estar separadas por una brecha de energías prohibidas que ningún electrón puede ocupar dentro del sólido. Veamos ahora cómo se usa este modelo para entender, digamos, las propiedades eléctricas de los sólidos. Para ello, apliquémoslo a dos casos particulares: el del sodio sólido y el del diamante. Cuando se forma un cristal de sodio, cada átomo contribuye con un electrón de valencia, que irá a acomodarse en los estados de una banda, buscando aquellos de menor energía. Según el principio de Paulí, en cada estado de la banda caben dos electrones, uno con el espín orientado en dirección contraria al otro, y no más. Pero entonces sólo se llena la mitad de estados de la capa, habiendo muchos estados disponibles, a los cuales puede llegar un electrón si se le excita un poco. Es decir, es fácil comunicarle energía cinética a uno de los electrones de valencia del cristal de sodio. Esto puede lograrse, por ejemplo, con un campo eléctrico. El sodio sólido es un buen conductor, como todos aquellos cristales que presentan en su estado de menor energía una banda semillena.
El caso del diamante es diferente. Ahora las dos bandas inferiores están completamente llenas, pues cada átomo contribuye con un número par de electrones de valencia. Entre los estados ocupados y los vacíos se encuentra una brecha de estados prohibidos, que en este caso particular es de 6 eV, cercana a la mitad de la energía de amarre de un electrón en el átomo de hidrógeno. Habría que comunicarle a un electrón al menos esta energía, para que pudiera moverse libremente. O sea, sujetarlo a un voltaje del orden de 6 voltios. Es muy difícil, y en consecuencia el diamante es un buen aislador. Obviamente, a medida que la brecha sea menor, el material es peor como aislante. Por ejemplo, la brecha en el caso del silicio es de sólo 1.1. eV. Esto se traduce en que el silicio sea mejor conductor que el diamante, aunque no tan bueno como un metal. Por ello se le ha clasificado como semiconductor.
IMPUREZAS Y SEMICONDUCTORES
Un sólido peculiar y muy útil sería aquél en que pudiéramos modificar la conductividad eléctrica a voluntad. De lo que hemos dicho, una manera de lograr esto seria cambiando el ancho de la brecha prohibida, o al menos logrando que haya algunos estados permitidos dentro de ella. Pensemos otra vez en el silicio, con sus cuatro electrones de valencia; de la red, cambiemos un átomo de silicio por otro de arsénico, que tiene cinco electrones en su capa más externa. De estos, cuatro van a intervenir en el enlace covalente con los átomos de silicio vecinos, pero el quinto electrón queda libre y puede ser itinerante en cuanto se le suministre un poco de energía. Este electrón puede transportar carga, con la consecuente modificación en la conductividad eléctrica. Otra posibilidad es, claro, cambiar un átomo de silicio por otro con sólo tres electrones de valencia. Entonces el nuevo átomo roba un electrón a alguno de sus vecinos, generando un agujero. Si este agujero se propaga, también lo hace una carga eléctrica positiva: se modifica, pues, la conductividad.
Lo que hemos hecho con el cristal perfecto de silicio es agregarle un defecto, que en este caso llamamos impureza. Agregando impurezas de manera controlada podemos generar materiales con diversas propiedades. Jugando con semiconductores impuros, como los descritos antes, fue como los físicos desarrollaron el transistor, que tanto ha influido en la vida del hombre contemporáneo. Si al semiconductor de silicio con arsénico le llamamos de tipo n, porque la conducción tiene lugar con una carga negativa, al otro le llamaremos de tipo p, ya que ahora se conduce carga moviendo un agujero, que implica falta de carga negativa o sea carga positiva. Un transistor consta de tres capas de semiconductores de uno y otro tipo. Por ejemplo, se tiene el transistor n-p-n, que consiste en una película muy delgada de semiconductor tipo p, emparedada entre dos capas de semiconductor tipo n. A la región central se le liaba base y a las capas exteriores se les denomina colector y emisor, respectivamente. Cuando el transistor opera, la base y el colector se conectan a la terminal negativa. Esto genera un flujo grande de electrones del emisor hacia la base, que por ser muy delgada no puede impedir el paso de todos los electrones, que entonces se difunden al colector dando lugar a una corriente a la salida de éste.
LA SUPERCONDUCTIVIDAD
Desde los tiempos de Romé de l'Isle hasta la era actual, que bien podría llamarse la de la microelectrónica, han transcurrido tan sólo dos siglos. Creemos ahora entender qué es un sólido y cómo modificar muchas de sus propiedades. En particular, hemos visto que es posible controlar la conductividad de muchos materiales y la consecuencia tecnológica tan impresionante que ello ha tenido. En nuestra discusión, el papel central lo han desempeñado los electrones, sin que aparentemente intervengan, salvo en forma incidental, los iones. Aunque de la estructura cristalina y del tipo de átomo surgen las características de las bandas, nunca hemos aquí considerado que los iones puedan también moverse. Sin embargo, es claro que los iones también se mueven, aunque lo hacen con menor celeridad que los electrones, ya que son miles de veces más pesados. Por otro lado, mientras el sólido permanezca como tal, los iones no podrán deambular por el cristal. Nuestra idea misma de un sólido, a diferencia de un líquido o de un gas, implica que los iones se encuentren limitados a la vecindad de su posición de equilibrio. Los iones, pues, sólo podrán oscilar con respecto a esos puntos de equilibrio. Y cuando la amplitud de la oscilación sea muy grande, comparable a la distancia interatómica en la red, el cristal empezará a fundirse.
Pensemos entonces en un modelo para las vibraciones de la red: los átomos se acoplan a sus vecinos próximos con algún tipo de resorte. Este es un modelo razonable, ya que los iones sólo vibran. Entre todos los posibles movimientos de los iones existen unos, llamados modos normales, en que todas las partículas oscilan con la misma frecuencia. A los cuantos de energía de estos osciladores normales se les conoce técnicamente con el nombre de fonones. Los fonones se parecen mucho a unas partículas microscópicas reales: tienen una velocidad y energías bien definidas; son del tipo bosón. Por otro lado, no pueden existir sin la malla, es decir, fuera del cristal. (Recuérdese que tal vez éste sea el caso de las partículas más fundamentales, los quarks.) Además, comunicando al sólido más energía, los átomos pueden vibrar más, creándose más excitación, o sea más fonones. Por otro lado, enfriando el cristal se logra que haya menos fonones. Por lo tanto, estas excitaciones de la malla se pueden crear y destruir. Los fonones son responsables de muchos fenómenos en los sólidos. En particular de la resistividad eléctrica, pues interfieren con los electrones. Como ya vimos, al bajar la temperatura los átomos se aquietan y desaparecen los fonones. Cuando bajamos mucho la temperatura, ya muy cerca del cero absoluto, puede darse que a traves de los iones un electrón atraiga a otro, venciendo la repulsión eléctrica entre las cargas negativas. Con esto, los dos electrones corren juntos y forman una pareja que, por estar formada de dos fermiones, se comporta como un bosón, que puede moverse sin resistencia a través del cristal. Estamos ahora frente a un superconductor!
Esta explicación de la superconductividad se dio apenas hace veinticinco años, aunque la observación de este fenómeno de bajas temperaturas data de principios de siglo. El estado superconductor es muy diferente al estado normal de los metales. Presenta una resistencia muy baja, no permite que un campo magnético penetre a su interior, en fin, representa toda una fase diferente. Dadas sus posibles aplicaciones: líneas de transmisión sin pérdida, electroimanes que pudieran generar campos magnéticos enormes, una gran cantidad de trabajo teórico y experimental se ha hecho en las últimas décadas para entender a los superconductores. Especial atención se ha puesto en buscar materiales que sean superconductores a la temperatura más alta posible, incluso a la temperatura ambiente. Aunque esto no se ha logrado todavía, si algún día ello fuera accesible, la superconductividad se convertiría sin duda en una de las principales tecnologías al alcance del hombre.
Nombre: Edymar Gonzalez A
C.I:19.502.773
http://www.reddeleducador.com.ar/una_ojeada_a_la_materia.htm
Podemos imaginarnos una infinidad de celdas unidad y, por tanto, de grecas o redes cristalinas resultantes. Sin embargo, las matemáticas han puesto cortapisas a nuestra imaginación pues el requisito de invariancia translacional es fuerte. Por ejemplo, las celdas unidad no pueden ser de forma pentagonal, ya que con pentágonos no es posible cubrir un plano totalmente sin dejar resquicios. Los geómetras han encontrado que sólo unos cuantos cientos de estructuras cristalinas diferentes son posibles y los han clasificado minuciosamente. Aunque ya los griegos habían hecho observaciones sobre los cristales, su estudio se inicia en el siglo XVII, por Stensen y Guglielmini, llegándose a encontrar en 1772 una ley enunciada por Romé de l'Isle: en todos los cristales de la misma substancia, los ángulos entre caras correspondientes tienen el mismo valor (cuando se les mide a igual temperatura). De l'Isle no logró proponer un modelo que diera cuenta de su observación y fue René JustHauy, al dejar caer un cristal de calcita y observar las caras idénticas de los fragmentos, quien sugirió la existencia de la celda unidad a la que llamó molécula integrante. Él empleaba sólo tres tipos distintos de moléculas integrantes pero, como ya lo mencionamos, se tienen muchos más como demostró Bravais el siglo pasado.
TEORÍA ATÓMICA DE LOS CRISTALES
Desde el punto de vista físico, cuál de esas estructuras cristalinas ha de tomar el sólido depende de varios factores, siendo los principales el tamaño de los átomos que lo forman y el tipo de fuerzas que actúa entre ellos. En función de estas últimas, y en completa analogía con el caso molecular, se clasifican los cristales en iónicos, covalentes o metálicos. Veamos algunas de sus características. Los cristales iónicos se enlazan con fuerzas interatómicas del tipo de Coulomb. Cuando un átomo con un electrón fuera de capa cerrada se encuentra con otro que requiere un electrón para completar la capa, el primero cede al electrón tan gustosamente como el otro lo atrapa. Pero entonces se forman dos iones, uno positivo porque cedió una carga negativa y el otro negativo porque adquirió un electrón adicional. Por lo tanto, se atraen eléctricamente con una fuerza omnidireccional, para la cual no existe dirección privilegiada. La descripción que hemos hecho se aplica a los átomos alcalinos frente a los halógenos. De hecho, los cristales iónicos por excelencia son los halogenuros alcalinos, siendo el más conocido el cloruro de sodio, la sal común.
A diferencia de los cristales iónicos, en que los electrones se reparten en los iones, en los cristales covalentes los electrones se comparten. La nube electrónica se dispone de manera tal que los iones positivos son atraídos hacia los sitios en que aquélla se localiza principalmente. Estos sitios preferenciales no son cualesquiera, por lo que el enlace covalente se da sólo en ciertas direcciones. Estos cristales covalentes surgen de elementos como el germanio, el silicio y el carbono. Por ejemplo, el carbono tiene cuatro electrones en su capa más externa, que se llena con ocho. Por lo tanto, al átomo de este elemento le da igual, por decirlo así, ceder sus electrones o aceptar otro número igual para llenar su capa y llegar a un estado de menor energía. Entonces, decide compartir sus cuatro electrones dando origen con un enlace covalente a un cristal tetraédrico.
Los cristales metálicos, finalmente, se dan cuando los átomos que los forman tienen electrones muy poco amarrados que con poca energía se liberan de su ión y prefieren deambular por todo el cristal. El origen de esta energía y en consecuencia la existencia del enlace metálico, se puede explicar con base en la mecánica cuántica y su principio de incertidumbre. Recordemos que este principio nos dice que no es posible definir al mismo tiempo la posición y la velocidad de una partícula. Entonces, mientras mayores sean las limitaciones espaciales a las cuales se restringe una partícula, también será más grande la incertidumbre en la velocidad, y por tanto la velocidad misma de la partícula. En otros términos, mientras menos intentemos localizar a un electrón, menor podrá ser su velocidad y, por lo tanto, su energía cinética. Esta disminución en energía que obtiene el electrón al moverse en todo el cristal y no dentro de un átomo, puede ser suficiente para compensar la energía de amarre al átomo. Esto sucede con los metales, en los cuales existe un gas de electrones, e inmersos en él los iones positivos. Estos últimos se atraen unos a los otros por medio del gas de electrones.
ALGUNAS PROPIEDADES DE LOS CRISTALES
Con la imagen que hemos construido de los tres tipos de cristales, ¿qué propiedades podemos esperar de cada uno? En el metal, pletórico de electrones itinerantes, casi libres, esperaríamos que un campo eléctrico pueda mover fácilmente a esas partículas de valencia no amarradas a ningún átomo en particular. Pero esto es lo que caracteriza a un buen conductor eléctrico. Por otro lado, cuando incide un rayo de luz sobre el cristal metálico, los campos electromagnéticos que forman la onda luminosa hacen oscilar a los electrones libres, que generan al acelerarse una radiación reflejada. Por lo tanto, conducen mal la luz al ser buenos reflectores: en otros términos, son opacos. En cuanto a sus propiedades térmicas, podemos esperar que los metales sean buenos conductores del calor, siendo la explicación microscópica semejante: si se aumenta la temperatura en un cierto punto del metal, esto implica aumentar la energía cinética de los electrones menos amarrados, que luego transportan esa energía a todos los confines del cristal. Por otro lado, el amarre metálico proveniente del gas de electrones es poco direccional. Los átomos de la red pueden desplazarse de su posición de mínima energía sin que ésta se altere mucho, ya que el gas de electrones, por decirlo así, se adapta a las nuevas posiciones de los átomos. Esto implica, claro, que los metales se pueden deformar sin llegar a la fractura. Finalmente, y otra vez debido a que el amarre entre iones procede del muy dúctil y poco exigente gas de electrones, se pueden dar muy diversas mezclas de dos metales, sin que dependan fuertemente de las proporciones relativas de cada elemento. De ahí que exista una gran variedad de aleaciones metálicas.
Pensemos ahora en la imagen nuestra de un cristal covalente, en que los electrones de valencia están ocupados en ligar al cristal. No hay en este caso quien transporte la carga o la energía térmica. Si no hay agentes portadores, la información no llega nunca, o al menos tarda mucho en llegar. Resultado neto, los cristales covalentes no son buenos conductores ni de la electricidad ni térmicos: se pueden usar como aislantes. Como dijimos antes, el enlace covalente es muy direccional y por tanto la posición de equilibrio en la red puede ser delicada. Estos sólidos, en consecuencia, no serán dúctiles sino quebradizos. Finalmente, no existe el mecanismo por el cual se reflejaba la luz al incidir en un metal, y podemos esperar que los cristales covalentes sean buenos conductores de la radiación electromagnética, o sea transparente.
Propiedades muy semejantes podemos esperar de los metales iónicos, si acaso alguna diferencia en sus propiedades ópticas y mecánicas. Por ejemplo, cuando incide sobre un cristal iónico una onda electromagnética cuya longitud es mucho mayor que la distancia típica que separa a los iones, tanto el ion positivo como el negativo sienten un campo eléctrico del mismo signo. Pero responden a él de manera contraria y se puede excitar una vibración en la malla cristalina si la frecuencia es la apropiada. A esta frecuencia, el cristal iónico absorbe la luz y ya no es transparente. ¿Quién no ha sentido frío en la mano al tocar una perilla metálica? ¿Quién no ha visto los destellos primorosos de los diamantes? ¿No sabemos que los mejores espejos están metalizados? ¿O que las extensiones eléctricas son en general de cobre, un metal? Todo ello se explica con los modelos para los sólidos que hemos desarrollado.
EL MODELO DE PARTÍCULAS INDEPENDIENTES
Así como en el átomo generamos un modelo de capas, en que sólo se toma en cuenta la atracción que ejerce el núcleo sobre cada electrón despreciando la interacción entre éstos; en la misma forma que para el núcleo inventamos un modelo de capas en que la parte principal de la interacción entre los nucleones se agota en producir una fuerza promedio; así también para los sólidos se ha desarrollado un modelo de partículas independientes que en este caso se llama la teoría de bandas. E igual que en la física atómica y en la teoría nuclear, este modelo resulta esencial para aplicar las ideas cuánticas al estudio de los cristales. Para entender la teoría de bandas, es conveniente imaginarse que formamos el cristal juntando un átomo tras otro. Por simplicidad supongamos que todos los átomos son iguales. Cuando los átomos están muy alejados unos de los otros, las energías de los electrones son idénticas a las del átomo aislado. Cuando los acercamos, la presencia de un átomo perturba un poco los niveles de energía del otro. En consecuencia, de cada nivel atómico se genera una banda de niveles, tan próximos entre sí que forman un continuo. En alguno de estos niveles de energía estarán los electrones del sólido.
Dependiendo de los átomos de que se trate, las bandas provenientes de niveles atómicos contiguos pueden superponerse o estar separadas por una brecha de energías prohibidas que ningún electrón puede ocupar dentro del sólido. Veamos ahora cómo se usa este modelo para entender, digamos, las propiedades eléctricas de los sólidos. Para ello, apliquémoslo a dos casos particulares: el del sodio sólido y el del diamante. Cuando se forma un cristal de sodio, cada átomo contribuye con un electrón de valencia, que irá a acomodarse en los estados de una banda, buscando aquellos de menor energía. Según el principio de Paulí, en cada estado de la banda caben dos electrones, uno con el espín orientado en dirección contraria al otro, y no más. Pero entonces sólo se llena la mitad de estados de la capa, habiendo muchos estados disponibles, a los cuales puede llegar un electrón si se le excita un poco. Es decir, es fácil comunicarle energía cinética a uno de los electrones de valencia del cristal de sodio. Esto puede lograrse, por ejemplo, con un campo eléctrico. El sodio sólido es un buen conductor, como todos aquellos cristales que presentan en su estado de menor energía una banda semillena.
El caso del diamante es diferente. Ahora las dos bandas inferiores están completamente llenas, pues cada átomo contribuye con un número par de electrones de valencia. Entre los estados ocupados y los vacíos se encuentra una brecha de estados prohibidos, que en este caso particular es de 6 eV, cercana a la mitad de la energía de amarre de un electrón en el átomo de hidrógeno. Habría que comunicarle a un electrón al menos esta energía, para que pudiera moverse libremente. O sea, sujetarlo a un voltaje del orden de 6 voltios. Es muy difícil, y en consecuencia el diamante es un buen aislador. Obviamente, a medida que la brecha sea menor, el material es peor como aislante. Por ejemplo, la brecha en el caso del silicio es de sólo 1.1. eV. Esto se traduce en que el silicio sea mejor conductor que el diamante, aunque no tan bueno como un metal. Por ello se le ha clasificado como semiconductor.
IMPUREZAS Y SEMICONDUCTORES
Un sólido peculiar y muy útil sería aquél en que pudiéramos modificar la conductividad eléctrica a voluntad. De lo que hemos dicho, una manera de lograr esto seria cambiando el ancho de la brecha prohibida, o al menos logrando que haya algunos estados permitidos dentro de ella. Pensemos otra vez en el silicio, con sus cuatro electrones de valencia; de la red, cambiemos un átomo de silicio por otro de arsénico, que tiene cinco electrones en su capa más externa. De estos, cuatro van a intervenir en el enlace covalente con los átomos de silicio vecinos, pero el quinto electrón queda libre y puede ser itinerante en cuanto se le suministre un poco de energía. Este electrón puede transportar carga, con la consecuente modificación en la conductividad eléctrica. Otra posibilidad es, claro, cambiar un átomo de silicio por otro con sólo tres electrones de valencia. Entonces el nuevo átomo roba un electrón a alguno de sus vecinos, generando un agujero. Si este agujero se propaga, también lo hace una carga eléctrica positiva: se modifica, pues, la conductividad.
Lo que hemos hecho con el cristal perfecto de silicio es agregarle un defecto, que en este caso llamamos impureza. Agregando impurezas de manera controlada podemos generar materiales con diversas propiedades. Jugando con semiconductores impuros, como los descritos antes, fue como los físicos desarrollaron el transistor, que tanto ha influido en la vida del hombre contemporáneo. Si al semiconductor de silicio con arsénico le llamamos de tipo n, porque la conducción tiene lugar con una carga negativa, al otro le llamaremos de tipo p, ya que ahora se conduce carga moviendo un agujero, que implica falta de carga negativa o sea carga positiva. Un transistor consta de tres capas de semiconductores de uno y otro tipo. Por ejemplo, se tiene el transistor n-p-n, que consiste en una película muy delgada de semiconductor tipo p, emparedada entre dos capas de semiconductor tipo n. A la región central se le liaba base y a las capas exteriores se les denomina colector y emisor, respectivamente. Cuando el transistor opera, la base y el colector se conectan a la terminal negativa. Esto genera un flujo grande de electrones del emisor hacia la base, que por ser muy delgada no puede impedir el paso de todos los electrones, que entonces se difunden al colector dando lugar a una corriente a la salida de éste.
LA SUPERCONDUCTIVIDAD
Desde los tiempos de Romé de l'Isle hasta la era actual, que bien podría llamarse la de la microelectrónica, han transcurrido tan sólo dos siglos. Creemos ahora entender qué es un sólido y cómo modificar muchas de sus propiedades. En particular, hemos visto que es posible controlar la conductividad de muchos materiales y la consecuencia tecnológica tan impresionante que ello ha tenido. En nuestra discusión, el papel central lo han desempeñado los electrones, sin que aparentemente intervengan, salvo en forma incidental, los iones. Aunque de la estructura cristalina y del tipo de átomo surgen las características de las bandas, nunca hemos aquí considerado que los iones puedan también moverse. Sin embargo, es claro que los iones también se mueven, aunque lo hacen con menor celeridad que los electrones, ya que son miles de veces más pesados. Por otro lado, mientras el sólido permanezca como tal, los iones no podrán deambular por el cristal. Nuestra idea misma de un sólido, a diferencia de un líquido o de un gas, implica que los iones se encuentren limitados a la vecindad de su posición de equilibrio. Los iones, pues, sólo podrán oscilar con respecto a esos puntos de equilibrio. Y cuando la amplitud de la oscilación sea muy grande, comparable a la distancia interatómica en la red, el cristal empezará a fundirse.
Pensemos entonces en un modelo para las vibraciones de la red: los átomos se acoplan a sus vecinos próximos con algún tipo de resorte. Este es un modelo razonable, ya que los iones sólo vibran. Entre todos los posibles movimientos de los iones existen unos, llamados modos normales, en que todas las partículas oscilan con la misma frecuencia. A los cuantos de energía de estos osciladores normales se les conoce técnicamente con el nombre de fonones. Los fonones se parecen mucho a unas partículas microscópicas reales: tienen una velocidad y energías bien definidas; son del tipo bosón. Por otro lado, no pueden existir sin la malla, es decir, fuera del cristal. (Recuérdese que tal vez éste sea el caso de las partículas más fundamentales, los quarks.) Además, comunicando al sólido más energía, los átomos pueden vibrar más, creándose más excitación, o sea más fonones. Por otro lado, enfriando el cristal se logra que haya menos fonones. Por lo tanto, estas excitaciones de la malla se pueden crear y destruir. Los fonones son responsables de muchos fenómenos en los sólidos. En particular de la resistividad eléctrica, pues interfieren con los electrones. Como ya vimos, al bajar la temperatura los átomos se aquietan y desaparecen los fonones. Cuando bajamos mucho la temperatura, ya muy cerca del cero absoluto, puede darse que a traves de los iones un electrón atraiga a otro, venciendo la repulsión eléctrica entre las cargas negativas. Con esto, los dos electrones corren juntos y forman una pareja que, por estar formada de dos fermiones, se comporta como un bosón, que puede moverse sin resistencia a través del cristal. Estamos ahora frente a un superconductor!
Esta explicación de la superconductividad se dio apenas hace veinticinco años, aunque la observación de este fenómeno de bajas temperaturas data de principios de siglo. El estado superconductor es muy diferente al estado normal de los metales. Presenta una resistencia muy baja, no permite que un campo magnético penetre a su interior, en fin, representa toda una fase diferente. Dadas sus posibles aplicaciones: líneas de transmisión sin pérdida, electroimanes que pudieran generar campos magnéticos enormes, una gran cantidad de trabajo teórico y experimental se ha hecho en las últimas décadas para entender a los superconductores. Especial atención se ha puesto en buscar materiales que sean superconductores a la temperatura más alta posible, incluso a la temperatura ambiente. Aunque esto no se ha logrado todavía, si algún día ello fuera accesible, la superconductividad se convertiría sin duda en una de las principales tecnologías al alcance del hombre.
Nombre: Edymar Gonzalez A
C.I:19.502.773
http://www.reddeleducador.com.ar/una_ojeada_a_la_materia.htm


 a
a b
b c
c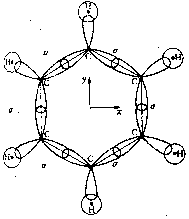 a
a  b
b